¿Qué es la Atrofia Muscular Espinal (SMA)?
Es una enfermedad genética neuronal, caracterizada por la pérdida de músculo esquelético causada por la progresiva degeneración de las células del asta anterior de la médula espinal.
Esta enfermedad causa debilidad y atrofia de los músculos voluntarios encargados de funciones tales como gatear, caminar, control del cuello y deglución. La debilidad ocurre más a menudo en la piernas que en los brazos. En consecuencia, el afectado no puede caminar, en muchos casos ni siquiera sentarse y, de acuerdo a la tipología, acarrea serios problemas respiratorios. Como característica también se observa que los pacientes con SMA son muy sociables e inteligentes.
La SMA es una enfermedad progresiva y está clasificada en Tipo I (Werding-Hoffman), Tipo II, Tipo III (Kugelberg – Welander) y Tipo IV (iniciación adulta). La tipología marca la gravedad de la enfermedad.
¿Que efectos causa la Atrofia Muscular Espinal (SMA)? |
| Según estadísticas efectuadas en Estados Unidos: • Uno de cada 6000 bebés nace con AME. • Una de cada 40 personas es portadora de los genes defectuosos. • El 50% de los niños afectados muere antes de llegar a los 2 años • No distingue raza ni sexo, etnia ni religión, es una enfermedad HUMANA. Según estudios realizados en Estados Unidos e información aportada por la Fundación Families of SMA, esta enfermedad es la principal causa de mortalidad infantil en niños menores de 2 años. |
¿Cómo se produce esta enfermedad? |
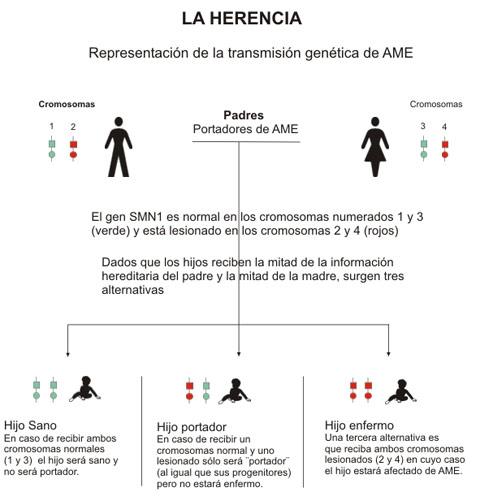
| Para que una persona esté afectada por esta enfermedad, debe ser concebida por dos personas portadoras del gen defectuoso, al momento de la concepción existe un 25% de probabilidades que se “unan” dos genes defectuosos de la pareja dando por resultado un afectado, el cual, hereda de sus padres genes que le producen fallas en el gen 1 de la supervivencia de las células motoneuronas (SMN1). Este Gen SMN1, es vital para la producción de una proteína que es esencial para el funcionamiento de las motoneuronas. Adicionalmente los afectados poseen una copia del gen pero incompleto, denominado SMN2, el cual no alcanza a producir el nivel de proteínas necesario para mover los músculos. A menor número de copias del gen, más grave es la tipología de la enfermedad. |