Què és la Atròfia Muscular Espinal (SMA)?
És una malaltia genètica neuronal, caracteritzada per la pèrdua de múscul esquelètic causada per la progressiva degeneració de les cèl·lules de l'asta anterior de la medul·la espinal.
Aquesta malaltia causa debilitat i atròfia dels músculs voluntaris encarregats de funcions com ara gatejar, caminar, control del coll i deglució. La debilitat passa més sovint en la cames que en els braços. En conseqüència, l'afectat no pot caminar, en molts casos ni tan sols seure i, d'acord amb la tipologia, comporta seriosos problemes respiratoris. Com a característica també s'observa que els pacients amb SMA són molt sociables i intel·ligents.
La SMA és una malaltia progressiva i està classificada en Tipus I (Werding-Hoffman), Tipus II, Tipus III (Kugelberg - Welander) i Tipus IV (iniciació adulta). La tipologia marca la gravetat de la malaltia.
¿Quins efectes causa la Atròfia Muscular Espinal (SMA)?
Segons estadístiques efectuades als Estats Units:
• Un de cada 6000 nadons neix amb AME.
• Una de cada 40 persones és portadora dels gens defectuosos.
• El 50% dels nens afectats mor abans d'arribar als 2 anys
• No distingeix raça ni sexe, ètnia ni religió, és una malaltia HUMANA.
Segons estudis realitzats als Estats Units i informació aportada per la Fundació Families of SMA, aquesta malaltia és la principal causa de mortalitat infantil en nens menors de 2 anys.
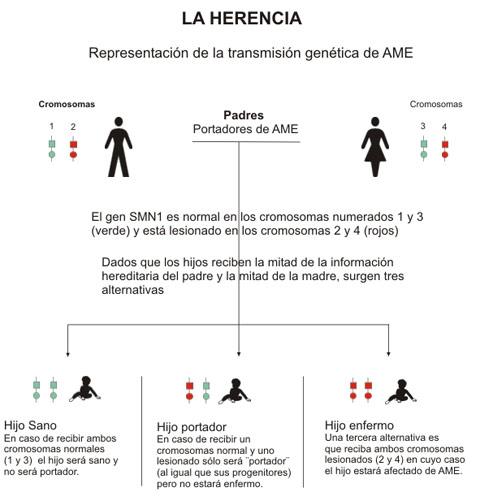
Com es produeix aquesta malaltia?
Perquè una persona estigui afectada per aquesta malaltia, ha de ser concebuda per dues persones portadores del gen defectuós, al moment de la concepció ha un 25% de probabilitats que es "s'uneixin" dos gens defectuosos de la parella donant per resultat un afectat, el qual, hereta dels seus pares gens que li produeixen falles en el gen 1 de la supervivència de les cèl·lules motoneurones (SMN1). Aquest Gen SMN1, és vital per a la producció d'una proteïna que és essencial per al funcionament de les motoneurones.
Addicionalment els afectats tenen una còpia del gen però incomplet, denominat SMN2, el qual no arriba a produir el nivell de proteïnes necessari per moure els músculs. A menor nombre de còpies del gen, més greu és la tipologia de la malaltia.